自免与炎症
代谢 / 肝脏疾病评价
肾脏疾病
眼科疾病
血液病
疼痛管理
耳科
中枢神经系统疾病
非人灵长类实验
>>填写表单后可查看
Alport 综合征是一种遗传性进行性肾炎,因 1927 年 Arthur C. Alport 首次系统描述而得名。它以肾脏损伤、听力下降、眼部异常三联征为典型表现,本质是编码Ⅳ型胶原的α3、α4、α5 链的COL4A3/COL4A4/COL4A5 基因发生突变,导致肾小球基底膜结构与功能缺陷,最终进展为终末期肾病。临床上,肾脏是主要受累器官,儿童期多以持续性血尿起病,逐渐出现蛋白尿,青壮年进展为终末期肾病,需透析或肾移植。听力损害多见于青少年,以高频听力下降为主,早期无症状、需检测发现,听力越差,肾衰风险越高。眼部特征性改变为前圆锥形晶状体与视网膜斑点病变,约 40% 患者早于蛋白尿出现,可辅助早期诊断。
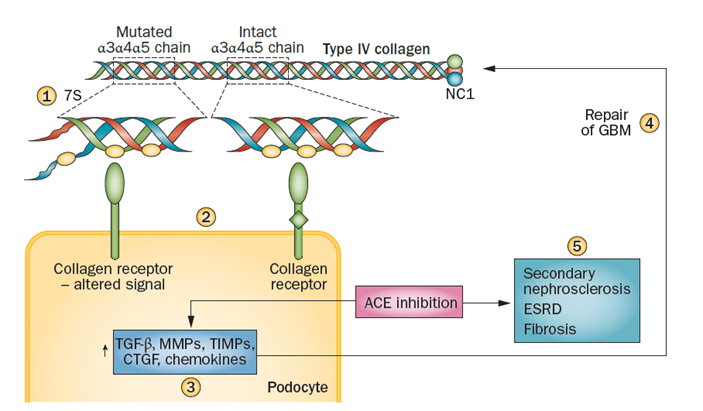
图1 - Alport 综合征的发病机制(DOI: 10.1038/nrneph.2012.259)
依据KDIGO 指南,Alport 综合征诊断以血尿、蛋白尿、慢性肾功能进展为核心,伴听力/眼部异常及阳性家族史;基因检测检出 COL4A3/4/5 致病变异为确诊金标准。典型病理特征为电镜下肾小球基底膜厚薄不均、致密层撕裂分层、呈篮网状改变,伴足突广泛融合,免疫荧光可见 Ⅳ 型胶原 α3/4/5 链表达缺失或异常。
Alport 综合征的临床前动物模型>>填写表单后可查看
|
Alport小鼠模型验证 |
Col4a5-R471X 小鼠采用 CRISPR/Cas9 精准引入人类 X 连锁 Alport 综合征常见致病无义突变 R471X,可高度模拟患者因 Ⅳ 型胶原α5 链缺陷导致的肾小球基底膜结构异常,临床相关性极强。该模型以 C57BL/6 近交系为背景,可稳定再现蛋白尿、肾功能衰退及肾小球基底膜病变等 Alport 综合征典型特征。模型病程进展规律,与人类疾病进程高度贴合,是 Alport 综合征临床转化研究的理想动物模型。
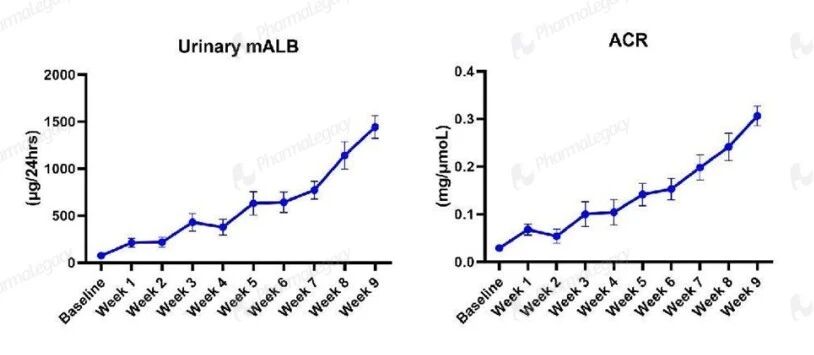
蛋白尿相关指标:尿微量白蛋白(mALB)及尿白蛋白/肌酐比值(ACR)自基线起呈持续、进行性升高,反映了 Alport 综合征由微量白蛋白尿向显性蛋白尿进展的典型过程,是肾损伤早期的敏感指标,并与临床诊断和随访的核心指标一致。
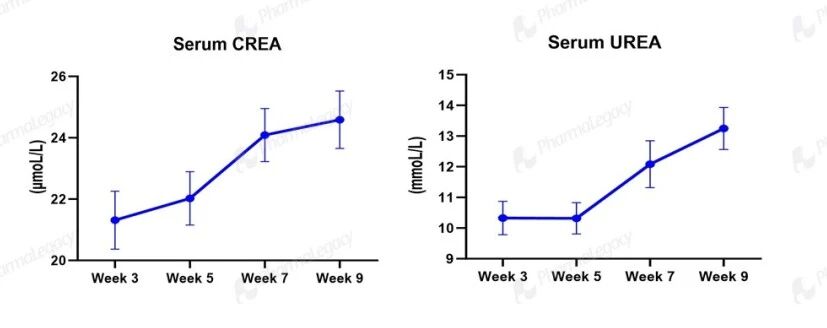
 肾功能血清学指标:血清肌酐(CREA)和尿素氮(UREA)随周龄逐渐升高;在 Alport 小鼠中,这一趋势再现了人类 Alport 综合征肾功能进行性衰退的临床特征,为肾保护药物评价提供了可靠依据。
肾功能血清学指标:血清肌酐(CREA)和尿素氮(UREA)随周龄逐渐升高;在 Alport 小鼠中,这一趋势再现了人类 Alport 综合征肾功能进行性衰退的临床特征,为肾保护药物评价提供了可靠依据。
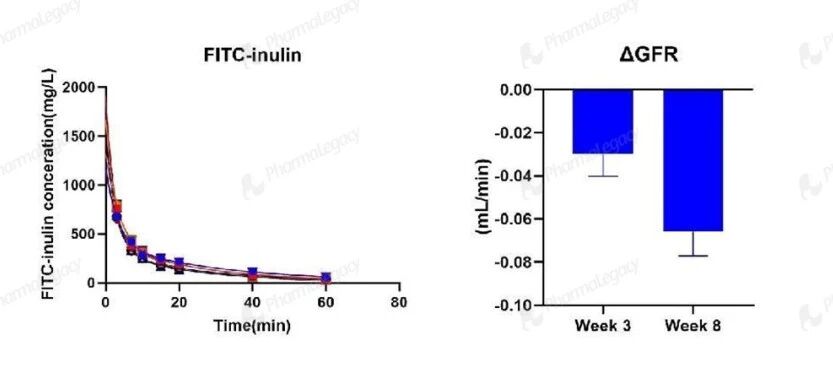
 肾小球滤过功能:FITC‑inulin 清除实验显示,GFR 自基线至 8 周显著、进行性下降,直接量化了 Alport 综合征中肾小球滤过功能的进行性减退;这一变化与临床 eGFR 持续下降的病程一致,可作为评价肾功能保护药物疗效的精准功能学终点。
肾小球滤过功能:FITC‑inulin 清除实验显示,GFR 自基线至 8 周显著、进行性下降,直接量化了 Alport 综合征中肾小球滤过功能的进行性减退;这一变化与临床 eGFR 持续下降的病程一致,可作为评价肾功能保护药物疗效的精准功能学终点。
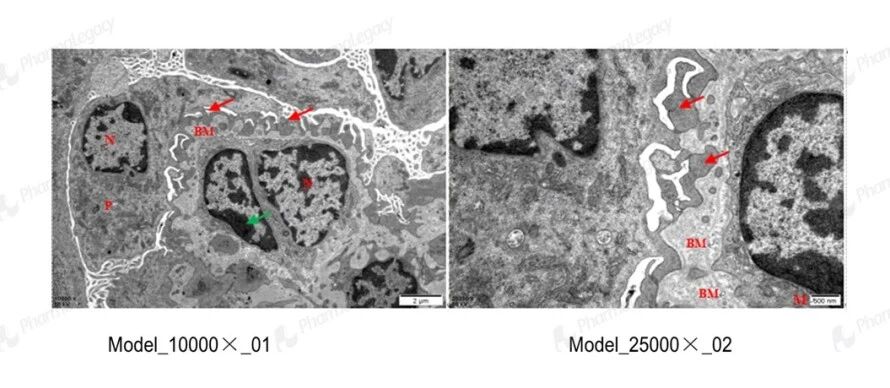
 肾组织电镜:Alport 小鼠肾组织电镜结果,高度还原了人类 X 连锁 Alport 综合征的特征性病理改变。电镜下可见肾小球毛细血管基底膜(GBM)显著增厚、厚薄不均、凹凸不平,完全复刻了 Alport 综合征标志性的 GBM 结构异常,是临床电镜诊断的核心依据;同时出现足细胞足突广泛融合、变短(红色箭头),直接对应患者蛋白尿发生的结构基础;系膜细胞核染色质边集(绿色箭头)则提示细胞活化,与肾纤维化、肾功能进行性衰退的临床病程高度吻合,为 Alport 综合征机制研究与药物评价提供了理想的病理模型支撑。
肾组织电镜:Alport 小鼠肾组织电镜结果,高度还原了人类 X 连锁 Alport 综合征的特征性病理改变。电镜下可见肾小球毛细血管基底膜(GBM)显著增厚、厚薄不均、凹凸不平,完全复刻了 Alport 综合征标志性的 GBM 结构异常,是临床电镜诊断的核心依据;同时出现足细胞足突广泛融合、变短(红色箭头),直接对应患者蛋白尿发生的结构基础;系膜细胞核染色质边集(绿色箭头)则提示细胞活化,与肾纤维化、肾功能进行性衰退的临床病程高度吻合,为 Alport 综合征机制研究与药物评价提供了理想的病理模型支撑。
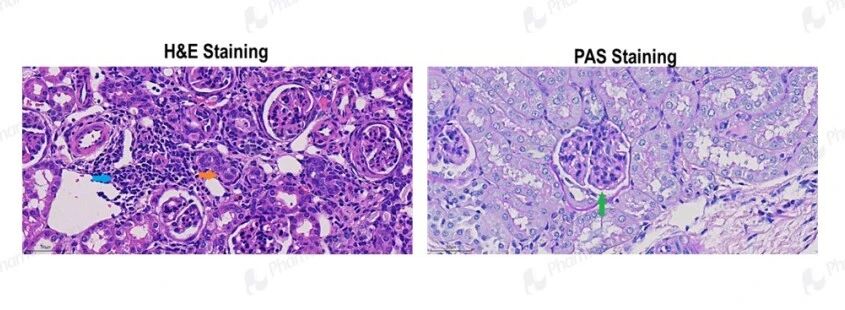
肾组织光镜:Alport 小鼠肾组织 H&E 与 PAS 染色结果,高度复刻了人类 Alport 综合征的典型光镜病理特征。H&E 染色可见明显炎症细胞浸润(蓝色箭头)、肾小管萎缩损伤(橙色箭头);PAS 染色清晰显示肾小球基底膜增厚(绿色箭头),与电镜超微结构改变相互印证。这些病理改变精准对应 Alport 综合征患者肾组织的炎症浸润、肾小管间质损伤及基底膜病变,与蛋白尿、肾功能进行性衰退的临床表现高度一致,为疾病机制研究与药物疗效评价提供了可靠的形态学依据。

在线咨询


请添加“业务小助手”企业微信


电话咨询

业务咨询
中国:
+86-130-0328-0039(业务咨询)
+86(21)6176-5100(总机)
Email:info@pharmalegacy.com
海外:
+1-617-803-9415(海外业务咨询)
+86-021-6176-5100*8051(海外业务咨询)
Email:info@pharmalegacy.com
投资者:
+86(21)6176-5100*8002
Email:IR@pharmalegacy.com

TOP