


2026-02-11
模型相关
从毒蜥唾液到糖尿病药物的GLP-1探索之旅
在美国西南部的沙漠中,栖息着一种名为吉拉毒蜥的生物。它拥有一种令人惊异的生理特性:在一次进食中可摄入相当于自身体重50%的食物,但其血糖水平却能保持异常稳定。这一现象引起了科学家的浓厚兴趣,并最终从其唾液中,发现了一把开启了当代代谢性疾病治疗革命的“神奇钥匙”。
1.人体的“代谢信使”:GLP-1的生理调控机制
在揭示这把“钥匙”的奥秘之前,我们首先需要理解人体内一个精密的生理调控系统。
餐后,分布于肠道黏膜的L细胞作为“化学感受器”被激活,释放出一种名为胰高血糖素样肽-1的激素,即 GLP-1。GLP-1可被视为一名高效的“代谢中枢信使”,它通过循环系统,向多个靶器官传递协调一致的指令:
这套多靶点调控机制,共同维持着机体的葡萄糖稳态与能量平衡。然而,GLP-1作为治疗药物存在一个根本性缺陷:极短的生物学半衰期。它分泌入血后,会迅速被广泛存在的二肽基肽酶-4(DPP-4)酶解失活,其活性半衰期不足2分钟。这就像一个信号发出后即刻中断,无法形成持续、稳定的治疗浓度。
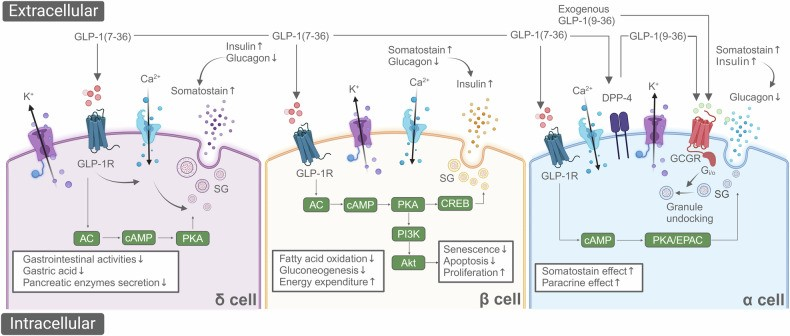
GLP-1在胰腺α、β和δ细胞中降低血糖的机制
DOI:10.1038/S41392-024-01931-Z
2.自然的启示:沙漠中的“长寿钥匙”
如何开发一种既能模拟GLP-1功能,又能抵抗体内快速降解的药物,成为代谢疾病治疗领域的关键挑战。科学家们将目光投向了自然界,最终在两种毒蜥的唾液中找到了答案。
研究者对墨西哥毒蜥和吉拉毒蜥的唾液进行了系统分析,从中分离出一类新型肽类物质,命名为exendin。对这一家族的深入研究,揭示了其结构与功能的精妙关系。
Exendin-3(源于墨西哥毒蜥)展现出复杂的受体交叉活性。在低浓度范围(0.1-10 nM)内,它能特异性激活一种新型的“exendin受体”,引起胰腺腺泡细胞内cAMP水平的剂量依赖性升高,但此浓度下不刺激淀粉酶分泌。然而,当浓度超过10 nM后,Exendin-3能进一步激活血管活性肠肽(VIP)受体,引发cAMP的二次升高并显著促进淀粉酶分泌,显示出对VIP受体信号通路的交叉激活能力。这一“exendin受体”,后续研究证实其主要对应哺乳动物中的GLP-1受体。
相比之下,Exendin-4(源于吉拉毒蜥)表现出更高的受体选择性。尽管与Exendin-3仅在N端第2、3位存在两个氨基酸差异(Gly²-Glu³取代了Ser²-Asp³),但这细微的结构变化使其药理特性发生根本改变。Exendin-4在整个浓度范围内(0.1-100 nM)仅专一性地作用于exendin受体,引起cAMP的单相饱和性升高(在0.1-10 nM达到平台),且完全不激活VIP受体或刺激淀粉酶分泌,也不干扰VIP与其受体的正常结合。
进一步研究阐明了exendin家族的结构-功能关系:这些肽的N端区域主要负责受体的激活和信号转导(诱导cAMP产生),而中段和C端区域则主导与受体的特异性结合。Exendin-4的N端微小变异,恰好优化了其与受体的相互作用界面,使其成为研究exendin受体的理想工具分子。
最关键的是,Exendin-4与人类GLP-1具有约53%的序列同源性,能有效激活人GLP-1受体,模拟GLP-1的生理效应,包括促进胰岛素分泌、抑制胰高血糖素释放、延缓胃排空等。更重要的是,其分子结构对二肽基肽酶-4的水解作用具有天然抵抗性,在人体内的半衰期可达数小时,与内源性GLP-1约2分钟的极短半衰期形成鲜明对比。
吉拉毒蜥能够一次性摄入大量食物而维持血糖稳定,其毒液中含有的Exendin-4可能是关键的适应性机制之一。这一自然界的分子优化,为后续开发长效GLP-1受体激动剂药物提供了至关重要的结构模板和设计思路。
3. 从生物唾液到治疗药物:“艾塞那肽”的诞生
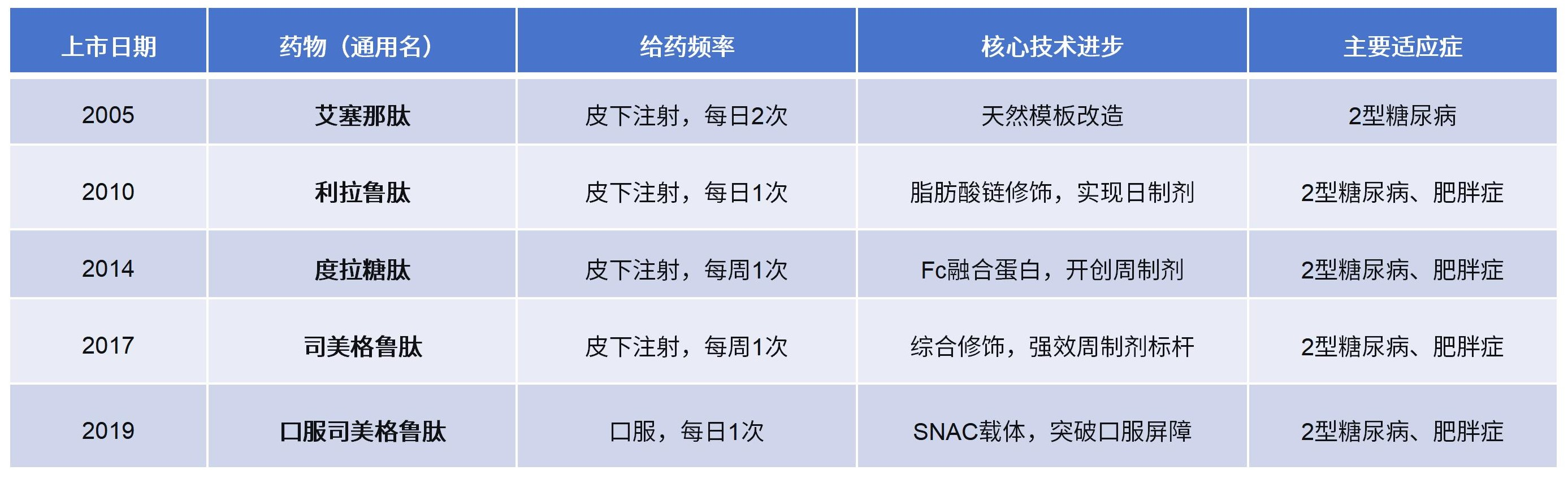
Exendin-4的发现,提供了设计GLP-1类药物的完美模板。经过多年的研发和临床试验,基于Exendin-4的人工合成药物——艾塞那肽,于2005年获得了美国FDA的批准,用于治疗2型糖尿病。它标志着第一代GLP-1受体激动剂的诞生。它证明了通过模拟并增强肠促胰岛素系统来治疗糖尿病,是一条完全可行的道路。它开启了“注射类肠促胰岛素”药物治疗的新时代。当然,作为初代产品,短效艾塞那肽仍有不足:它需要患者每日注射两次,且降糖和减重效果仍有提升空间。
GLP-1RA:从注射到口服的药物革命进化
第一把“钥匙”艾塞那肽的成功,点燃了药学界改造GLP-1的热情。一场围绕“长效、强效、便利”的分子设计竞赛悄然打响。这不仅是技术的迭代,更是人类智慧对生理密码的深度破译与重塑。而每一个里程碑式药物的诞生,从体外分子的设计到临床前功效的确证,都离不开模型验证,这是创新分子从“概念”转化为“药物”不可或缺的基石
4. 目标明确:攻克“短效”与“不便”
初代GLP-1受体激动剂(GLP-1RA)艾塞那肽的短板很明显:半衰期约2.4小时,需每日注射两次。这不仅给患者带来不便,也影响了血糖的平稳控制。
因此,药化学家的目标非常清晰:
实现这些目标,核心在于对抗体内的两大清除机制:① 二肽基肽酶-4(DPP-4酶)的降解;② 肾脏的快速过滤排泄。
5. 技术路径:四大“改造术”
为了实现这些目标,科学家们主要研发了四种“升级”技术,让GLP-1药物变得更强、更持久、更方便。
5.1. 挂上“救生圈”:脂肪酸修饰术(利拉鲁肽)
设计原理:在GLP-1类似物分子(利拉鲁肽与天然GLP-1同源性达97%)的第26位赖氨酸上,通过一个谷氨酸 spacer,共价连接一个16碳的棕榈酸脂肪酸侧链,就像给它系上了一个“救生圈”。
作用机制:这个脂肪“救生圈”能让药物牢牢抓住血液里大量存在的“运输船”——人血清白蛋白(HAS)。白蛋白分子量大(约66.5 kDa),不会被肾脏轻易过滤掉,于是药物就能搭着这艘“大船”在血液里长时间巡航,避免了被快速清除。同时,这种结合也部分保护了药物不被降解酶(DPP-4)“剪断”。
成果:利拉鲁肽的半衰期延长至13小时,实现每日一次皮下注射,并在肥胖症治疗领域获得批准,证明了GLP-1类药物在体重管理上的巨大潜力。
5.2. 穿上“循环盔甲”:Fc融合蛋白技术(度拉糖肽)
设计原理:将两个经过氨基酸修饰(第8位为甘氨酸以提高DPP-4抗性)的GLP-1类似物肽链,通过一个柔性链接肽,分别融合到人免疫球蛋白G4 的Fc片段上,形成同源二聚体。
作用机制:这是主动对抗清除的典范。Fc片段本身可通过与新生儿Fc受体(FcRn)的相互作用,启动“循环利用”机制,避免被溶酶体降解,天然具有长半衰期(约21天)。融合后,整个分子量远超肾小球滤过阈值(>70 kDa),完全避免了肾脏清除。此外,大分子结构也有效抵抗了酶解。
成果:度拉糖肽平均半衰期达到约5天,开创了每周一次给药的“周制剂”时代,让治疗变得更加便捷。
5.3. “精装修”强化:综合化学优化(司美格鲁肽)
设计原理:这是“改造术”的集大成者。
成果:半衰期进一步延长至约7天,成为强效周制剂的标杆。卓越的减重效果更是将这类药物的应用从将GLP-1RA的应用推向了新的高峰,彻底确立了其在肥胖症治疗中的核心地位。
5.4. 实现“口服梦”:胃部吸收增强技术(口服司美格鲁肽)
核心挑战:肽类药物想口服,必须闯过胃酸的腐蚀、肠道酶的消化、以及穿透肠黏膜吸收三大关,难度极高。其根源在于司美格鲁肽作为大分子多肽,在胃肠道中极易被蛋白酶降解,且难以被动扩散通过肠上皮屏障,这是其口服生物利用度极低的根本原因。
技术原理:将司美格鲁肽与一种叫做N-[8-(2-羟基苯甲酰基)氨基辛酸钠] (SNAC)的吸收增强剂制成片剂:SNAC在胃里形成一个临时的“保护罩”,能局部、短暂地提升胃内pH值,保护药物不被胃酸快速破坏。同时,它能促进药物穿透胃黏膜细胞,直接进入血液循环。
成果与局限:2019年,口服司美格鲁肽成功上市。虽然最终能被吸收进血液的药量比例很低(生物利用度约0.4%-1%),远低于注射剂,但通过提高药片中的剂量(最高14毫克/天),血液中的药物浓度足以达到有效治疗水平。这实现了每日一次口服的历史性突破,满足了部分患者对无针治疗的迫切需求。
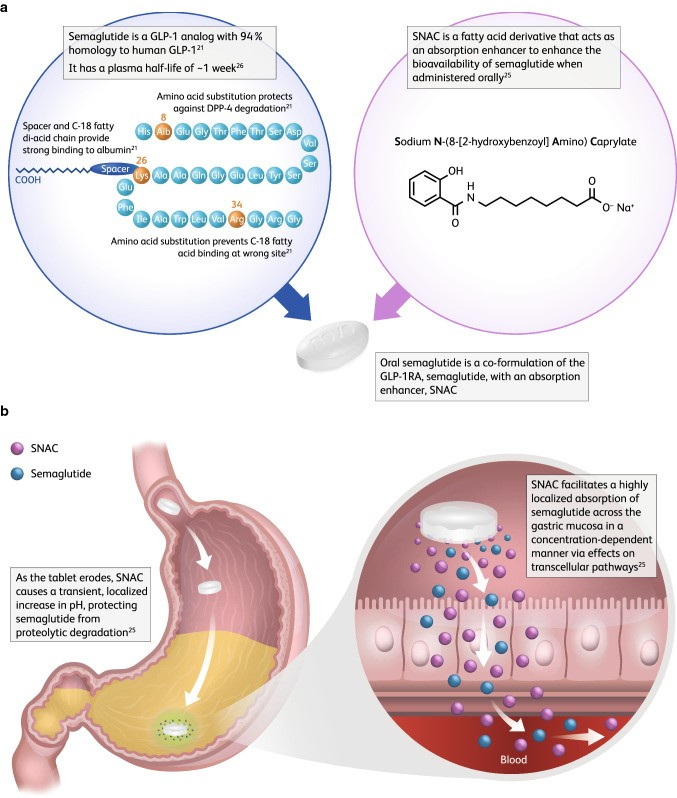
(a)司美格鲁肽与SNAC的结构(b)以及SNAC促进司美格鲁肽吸收的作用机制
doi: 10.1007/s40265-021-01499-w
澎立生物代谢疾病评价平台
从毒蜥唾液的偶然发现,到最终成为造福亿万患者的革命性药物,GLP-1激动剂的研发历程清晰地揭示了:基础科学的洞见是起点,而成功的转化离不开精准、可靠的临床前研究模型的系统验证。药物的有效性及作用机制,需要在进入人体试验前,在模拟人类疾病的动物模型中得到充分的评估。而这一关键验证环节,正是澎立生物服务平台的核心定位与价值体现,为药物临床前有效性及作用机制评估提供核心支撑。
以2型糖尿病的药物研发为例,澎立生物代谢性疾病平台构建了全面且具有高度临床相关性的糖尿病/肥胖动物模型体系,为GLP-1类乃至其他新型降糖/减重药物的开发提供了关键的“试验场”和“加速器”:
利用这些模型,澎立生物平台能够为客户提供综合性的临床前药效评价服务。例如,通过监测模型动物的空腹/随机血糖、糖耐量(OGTT/IPGTT)、胰岛素水平、糖化血红蛋白(HbA1c)等关键指标,精准量化药物的降糖效果;通过体成分分析(如 DEXA、MRI)评估减重及体脂分布改善情况;通过组织病理学、分子生物学及多组学分析,深入阐释药物的作用靶点与通路机制。
澎立生物案例:
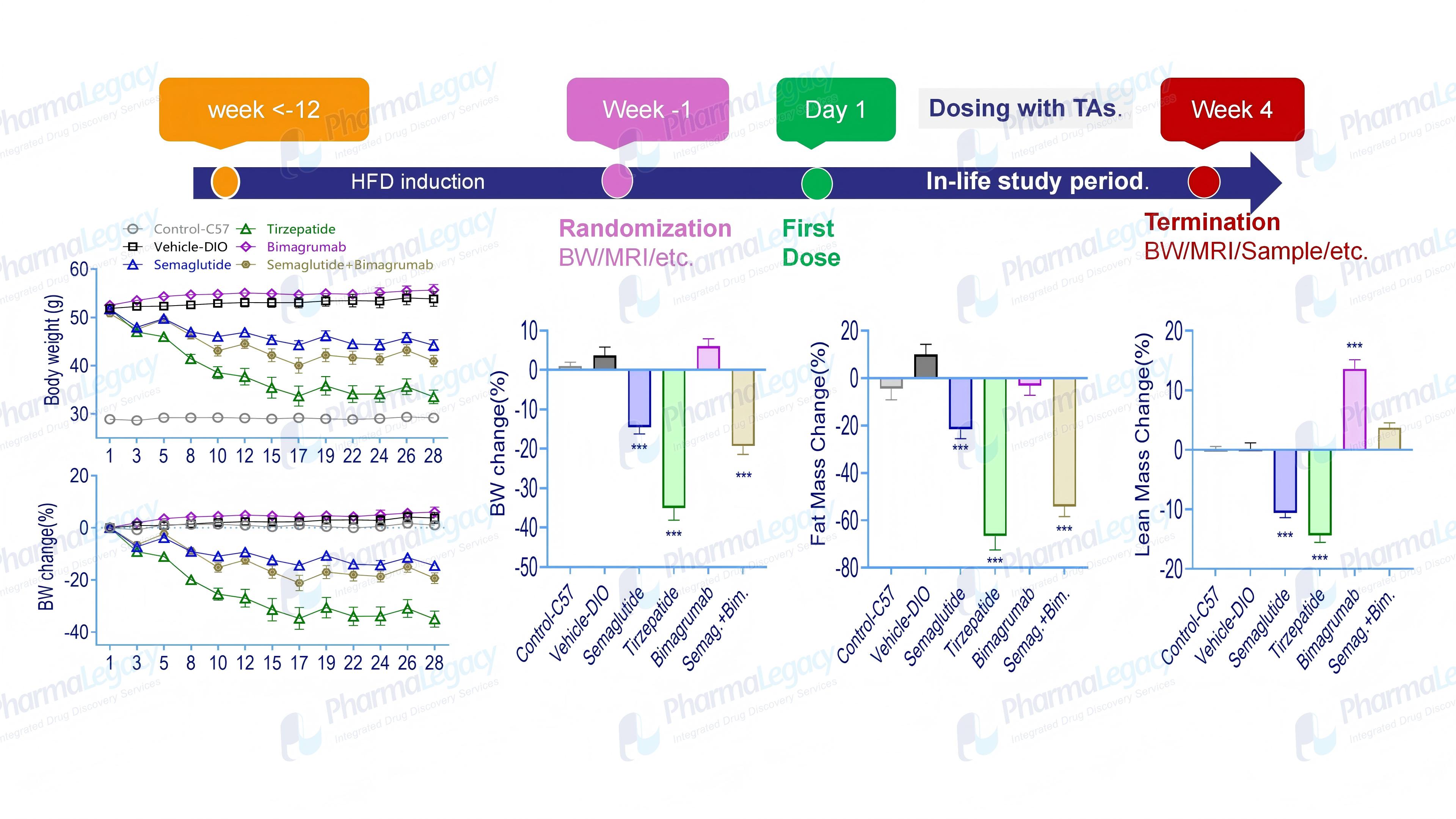
T2D模型
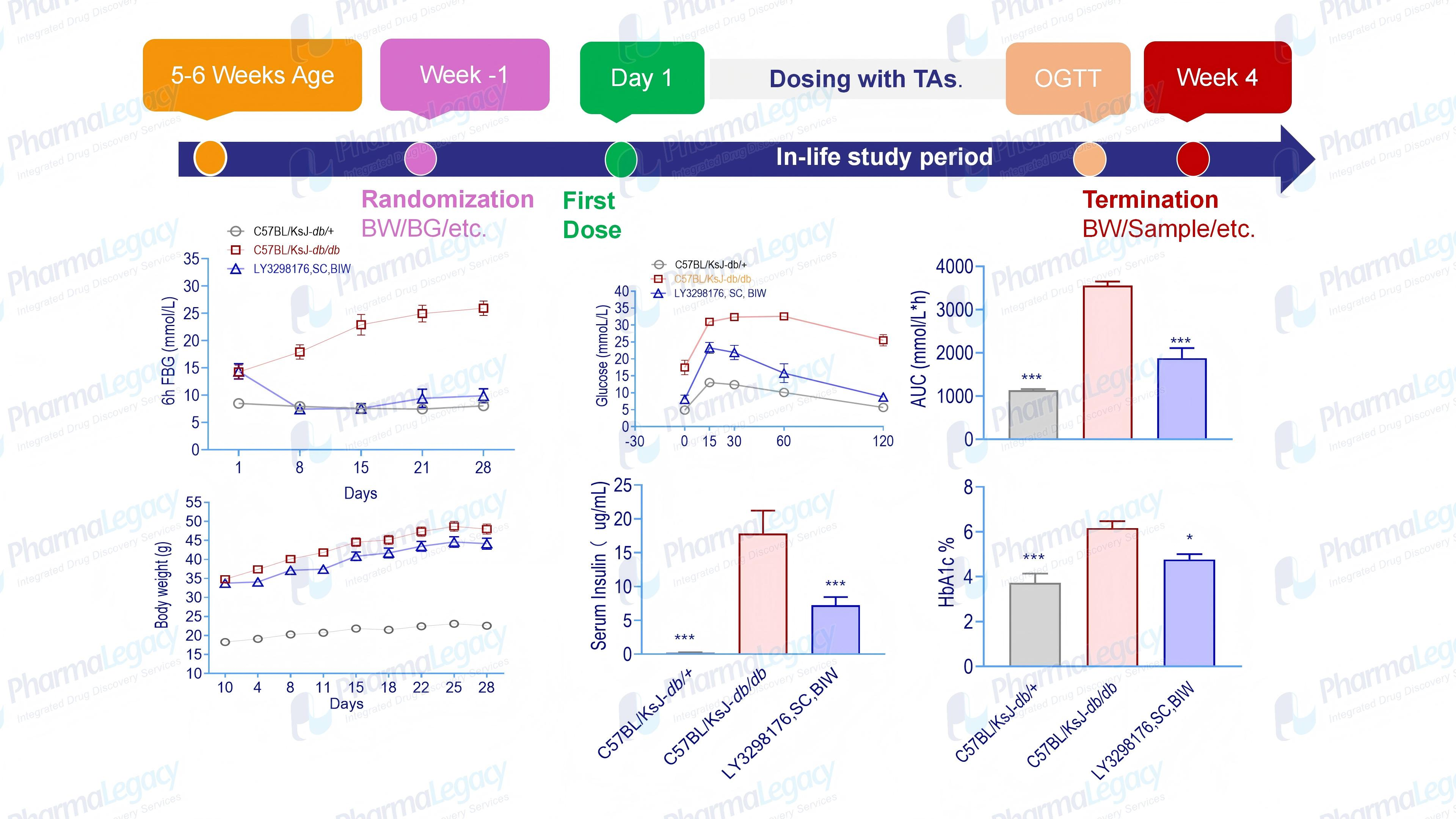
肥胖模型案例
参考文献
[1] Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem. 1992 Apr 15;267(11):7402-5.
[2] Bell GI, Santerre RF, Mullenbach GT. Hamster preproglucagon contains the sequence of glucagon and two related peptides. Nature. 1983 Apr 21;302(5910):716-8.
[3] Bell GI, Sanchez-Pescador R, Laybourn PJ, Najarian RC. Exon duplication and divergence in the human preproglucagon gene. Nature. 1983 Jul 28-Aug 3;304(5924):368-71.
[4] Zhao X, Wang M, Wen Z, Lu Z, Cui L, Fu C, Xue H, Liu Y, Zhang Y. GLP-1 Receptor Agonists: Beyond Their Pancreatic Effects. Front Endocrinol (Lausanne). 2021 Aug 23;12:721135.
[5] Christel CM, DeNardo DF, Secor SM. Metabolic and digestive response to food ingestion in a binge-feeding lizard, the Gila monster (Heloderma suspectum). J Exp Biol. 2007 Oct;210(Pt 19):3430-9.
[6] Furman BL. The development of Byetta (exenatide) from the venom of the Gila monster as an anti-diabetic agent. Toxicon. 2012 Mar 15;59(4):464-71.
[7] Zheng Z, Zong Y, Ma Y, Tian Y, Pang Y, Zhang C, Gao J. Glucagon-like peptide-1 receptor: mechanisms and advances in therapy. Signal Transduct Target Ther. 2024 Sep 18;9(1):234.
[8] Knop FK, Brønden A, Vilsbøll T. Exenatide: pharmacokinetics, clinical use, and future directions. Expert Opin Pharmacother. 2017 Apr;18(6):555-571.
[9] Drucker DJ, Dritselis A, Kirkpatrick P. Liraglutide. Nat Rev Drug Discov. 2010 Apr;9(4):267-8.
[10] Sanford M. Dulaglutide: first global approval. Drugs. 2014 Nov;74(17):2097-103.
[11] Tibble CA, Cavaiola TS, Henry RR. Longer acting GLP-1 receptor agonists and the potential for improved cardiovascular outcomes: a review of current literature. Expert Rev Endocrinol Metab. 2013 May;8(3):247-259.
[12] Chang KC, Shao SC, Kuo S, Yang CY, Chen HY, Chan YY, Ou HT. Comparative effectiveness of dulaglutide versus liraglutide in Asian type 2 diabetes patients: a multi-institutional cohort study and meta-analysis. Cardiovasc Diabetol. 2020 Oct 9;19(1):172.
[13] Dhillon S. Semaglutide: First Global Approval. Drugs. 2018 Feb;78(2):275-284.
[14] Andersen A, Knop FK, Vilsbøll T. A Pharmacological and Clinical Overview of Oral Semaglutide for the Treatment of Type 2 Diabetes. Drugs. 2021 Jun;81(9):1003-1030.
[15] Rasmussen MF. The development of oral semaglutide, an oral GLP-1 analog, for the treatment of type 2 diabetes. Diabetol Int. 2020 Jan 4;11(2):76-86.
[16] Rosenstock J, Frias J, Jastreboff AM, Du Y, Lou J, Gurbuz S, Thomas MK, Hartman ML, Haupt A, Milicevic Z, Coskun T. Retatrutide, a GIP, GLP-1 and glucagon receptor agonist, for people with type 2 diabetes: a randomised, double-blind, placebo and active-controlled, parallel-group, phase 2 trial conducted in the USA. Lancet. 2023 Aug 12;402(10401):529-544.

在线咨询


请添加“业务小助手”企业微信


电话咨询

业务咨询
中国:
+86-130-0328-0039(业务咨询)
+86(21)6176-5100(总机)
Email:info@pharmalegacy.com
海外:
+1-617-803-9415(海外业务咨询)
+86-021-6176-5100*8051(海外业务咨询)
Email:info@pharmalegacy.com
投资者:
+86(21)6176-5100*8002
Email:IR@pharmalegacy.com

TOP






 返回列表
返回列表










