


2026-04-27
模型相关
Alport综合征病理特征及临床诊断
Alport 综合征是一种遗传性进行性肾炎,因 1927 年 Arthur C. Alport 首次系统描述而得名。它以肾脏损伤、听力下降、眼部异常三联征为典型表现,本质是编码Ⅳ型胶原的α3、α4、α5 链的COL4A3/COL4A4/COL4A5 基因发生突变,导致肾小球基底膜结构与功能缺陷,最终进展为终末期肾病。临床上,肾脏是主要受累器官,儿童期多以持续性血尿起病,逐渐出现蛋白尿,青壮年进展为终末期肾病,需透析或肾移植。听力损害多见于青少年,以高频听力下降为主,早期无症状、需检测发现,听力越差,肾衰风险越高。眼部特征性改变为前圆锥形晶状体与视网膜斑点病变,约 40% 患者早于蛋白尿出现,可辅助早期诊断。
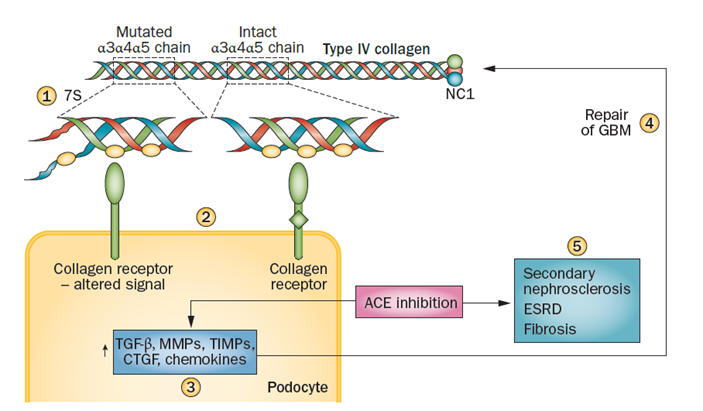
图1 - Alport 综合征的发病机制[1]
依据KDIGO 指南,Alport 综合征诊断以血尿、蛋白尿、慢性肾功能进展为核心,伴听力/眼部异常及阳性家族史;基因检测检出 COL4A3/4/5 致病变异为确诊金标准。典型病理特征为电镜下肾小球基底膜厚薄不均、致密层撕裂分层、呈篮网状改变,伴足突广泛融合,免疫荧光可见 Ⅳ 型胶原 α3/4/5 链表达缺失或异常。
Alport肾病综合征的标准治疗与创新疗法研究
1、标准治疗
Alport 综合征临床标准治疗以RAAS 阻断剂(ACEI/ARB)为一线核心,是唯一公认可显著延缓肾衰的方案。患者需从儿童期尽早启动。血管紧张素转换酶抑制剂(ACEI,如雷米普利)与血管紧张素 Ⅱ 受体拮抗剂(ARB,如氯沙坦)可显著减少蛋白尿、减轻肾纤维化,推迟终末期肾病(ESRD)发生。
SGLT2 抑制剂
SGLT2 抑制剂最初用于 2 型糖尿病,通过抑制近端肾小管葡萄糖重吸收发挥降糖作用。其可增强管球反馈、收缩入球小动脉,降低肾小球内高压;与 RAAS 抑制剂介导的出球小动脉扩张形成协同,进一步减轻肾小球压力。此外,该药还能减少钠重吸收、降低血容量,并抑制肾脏炎症与纤维化,带来多重肾保护获益。基于明确的肾脏与心血管获益及良好安全性,2024 年欧洲三大肾病学会联合指南推荐,对合并蛋白尿与慢性肾脏病的成年 Alport 综合征患者使用 SGLT2 抑制剂。
内皮素受体阻断剂
斯帕森坦作为内皮素A型受体与血管紧张素Ⅱ1型受体的双重拮抗剂,可同时阻断两条关键致肾损伤通路。临床前研究显示其可显著降低蛋白尿、延缓肾功能进展,并对听力异常具有一定改善作用。临床方面,除已在IgA肾病中显示明确降蛋白尿效果外,正在进行的Ⅱ期EPPIK研究纳入包括Alport综合征在内的蛋白尿性肾小球疾病患者,以尿蛋白/肌酐比变化为主要终点,评估其肾脏保护作用。
阿曲生坦为选择性内皮素A型受体拮抗剂,已在IgA肾病中获批。在糖尿病肾病研究中证实其可降低蛋白尿并减少肾脏不良结局风险。Ⅱ期AFFINITY研究已纳入Alport综合征患者,初步结果提示其可进一步降低蛋白尿,潜在具有延缓疾病进展的作用,但尚缺乏确证性结局数据。安全性方面,需关注水肿和贫血等与内皮素通路抑制相关的不良反应。
降脂与代谢调控药物
R3R01 作为新型口服降脂小分子,可上调 ABCA1 转运体表达,促进肾细胞内多余胆固醇排出,减轻脂质沉积引发的足细胞损伤与间质纤维化,Ⅱ 期临床招募 12 岁以上、尿蛋白 / 肌酐比≥1g/g 的 X 连锁或常染色体隐性 Alport 患者,主要观察蛋白尿变化。
二甲双胍通过抗炎、抗纤维化延缓肾损伤,在动物实验中可改善肾功能、延长生存期,中国已启动针对 10–17 岁 X 连锁及常染色体隐性患者的临床研究,以 eGFR 与蛋白尿为主要终点,评估非糖尿病患者中的安全性与肾保护效果。
羟氯喹
羟氯喹凭借免疫调节作用进入临床探索。该药可抑制 Toll 样受体、减少炎症因子释放,在 IgA 肾病中已展现降蛋白尿作用且安全性优于糖皮质激素。中国开展的 Ⅱ 期 CHXLAS 研究纳入 50 例儿童患者,在稳定 RAAS 抑制基础上联合羟氯喹,以尿红细胞计数为主要终点、蛋白尿与 eGFR 为次要终点,验证其对 Alport 综合征血尿、蛋白尿的控制效果,为免疫因素参与的病情进展提供新干预思路。
未来探索方向
Alport 综合征的未来治疗正逐步从传统对症治疗迈向分子与基因层面的精准干预。当前探索方向包括干细胞移植、无义突变通读治疗、外显子跳跃、分子伴侣、蛋白替代以及基因编辑等。其中,ELX-02等通读药物已进入Ⅱ期临床,外显子跳跃和分子伴侣在特定突变类型中展现出潜力,CRISPR/Cas9基因编辑及X染色体再激活等策略亦取得早期研究进展。然而,多数疗法仍处于实验或早期临床阶段,疗效与安全性仍需进一步验证,距离广泛临床应用尚有一定距离。
Alport 综合征的临床前动物模型
澎立生物案例:Alport小鼠模型验证
Col4a5-R471X 小鼠采用 CRISPR/Cas9 精准引入人类 X 连锁 Alport 综合征常见致病无义突变 R471X,可高度模拟患者因 Ⅳ 型胶原α5 链缺陷导致的肾小球基底膜结构异常,临床相关性极强。该模型以 C57BL/6 近交系为背景,可稳定再现蛋白尿、肾功能衰退及肾小球基底膜病变等 Alport 综合征典型特征。模型病程进展规律,与人类疾病进程高度贴合,是 Alport 综合征临床转化研究的理想动物模型。
蛋白尿相关指标:尿微量白蛋白(mALB)及尿白蛋白/肌酐比值(ACR)自基线起呈持续、进行性升高,反映了 Alport 综合征由微量白蛋白尿向显性蛋白尿进展的典型过程,是肾损伤早期的敏感指标,并与临床诊断和随访的核心指标一致。
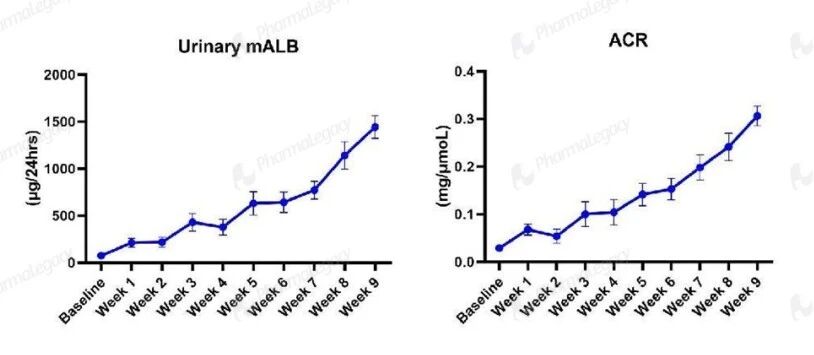
肾功能血清学指标:血清肌酐(CREA)和尿素氮(UREA)随周龄逐渐升高;在 Alport 小鼠中,这一趋势再现了人类 Alport 综合征肾功能进行性衰退的临床特征,为肾保护药物评价提供了可靠依据。
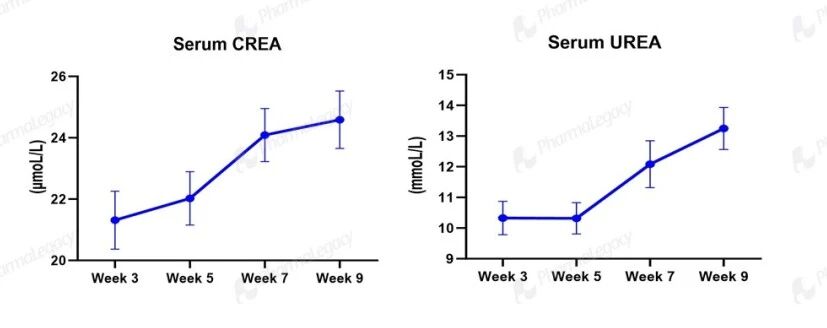
肾小球滤过功能:FITC‑inulin 清除实验显示,GFR 自基线至 8 周显著、进行性下降,直接量化了 Alport 综合征中肾小球滤过功能的进行性减退;这一变化与临床 eGFR 持续下降的病程一致,可作为评价肾功能保护药物疗效的精准功能学终点。
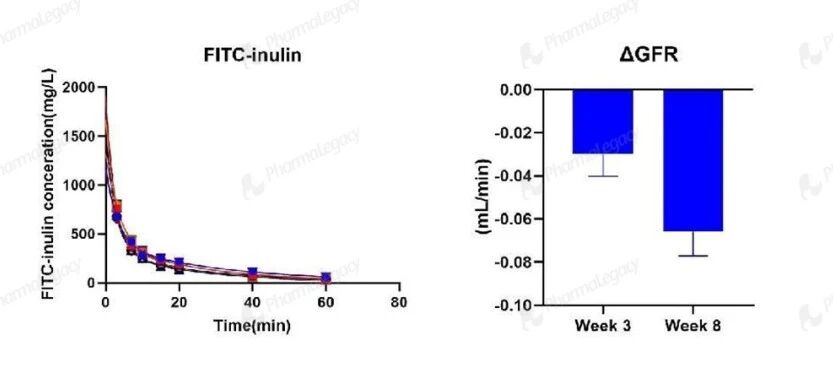
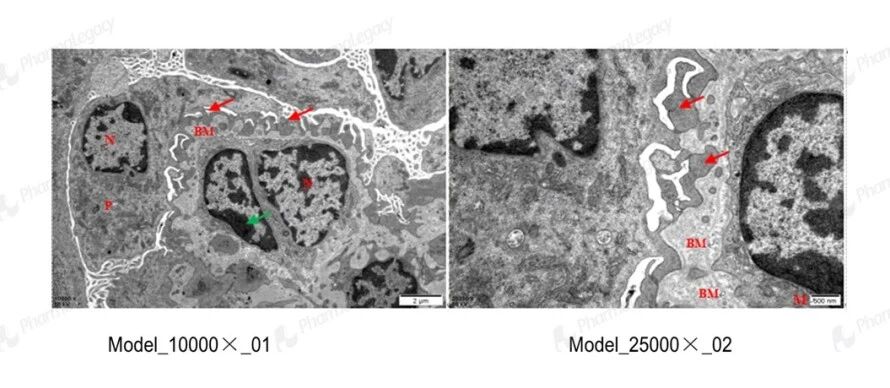
肾组织电镜:Alport 小鼠肾组织电镜结果,高度还原了人类 X 连锁 Alport 综合征的特征性病理改变。电镜下可见肾小球毛细血管基底膜(GBM)显著增厚、厚薄不均、凹凸不平,完全复刻了 Alport 综合征标志性的 GBM 结构异常,是临床电镜诊断的核心依据;同时出现足细胞足突广泛融合、变短(红色箭头),直接对应患者蛋白尿发生的结构基础;系膜细胞核染色质边集(绿色箭头)则提示细胞活化,与肾纤维化、肾功能进行性衰退的临床病程高度吻合,为 Alport 综合征机制研究与药物评价提供了理想的病理模型支撑。
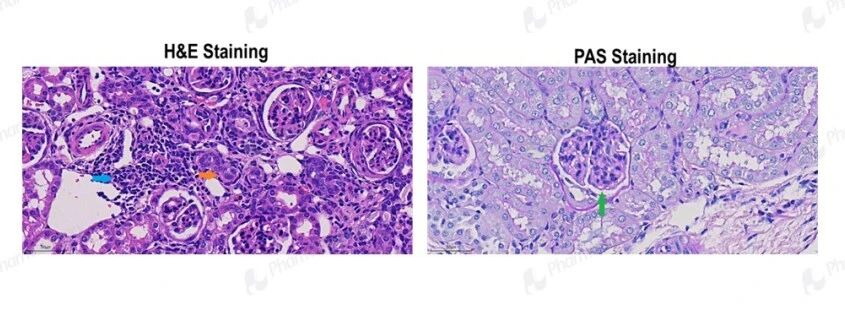
肾组织光镜:Alport 小鼠肾组织 H&E 与 PAS 染色结果,高度复刻了人类 Alport 综合征的典型光镜病理特征。H&E 染色可见明显炎症细胞浸润(蓝色箭头)、肾小管萎缩损伤(橙色箭头);PAS 染色清晰显示肾小球基底膜增厚(绿色箭头),与电镜超微结构改变相互印证。这些病理改变精准对应 Alport 综合征患者肾组织的炎症浸润、肾小管间质损伤及基底膜病变,与蛋白尿、肾功能进行性衰退的临床表现高度一致,为疾病机制研究与药物疗效评价提供了可靠的形态学依据。
总结
近年来,Alport 综合征的诊疗领域正不断迎来突破性进展,新型靶点发掘与创新治疗策略的探索,正为这部分罕见肾病患者点亮长期获益的希望之光。而可靠的临床前模型,正是串联基础研究与临床转化的核心桥梁。澎立生物肾病研究平台深度聚焦领域核心需求,成功构建了一系列肾病动物模型,不仅能精准复现人类 Alport 综合征的病理特征与关键生化表型,还可根据研发场景灵活定制个性化方案。从疾病机制的深度解析,到候选药物的高通量筛选、精准药效验证,平台都能提供全流程的坚实支撑。未来,我们将以专业的模型构建技术与一体化平台能力,携手药物研发伙伴,加速推动突破性疗法的临床转化,尽早惠及更多饱受病痛折磨的患者。
参考文献:
[1] Kruegel J, Rubel D, Gross O. Alport syndrome-insights from basic and clinical research. Nat Rev Nephrol. 2013 Mar;9(3):170-8.
[2] Rheault MN. Treatment Approaches for Alport Syndrome. J Am Soc Nephrol. 2026 Jan 1;37(1):172-179.
[3] Gross O, Boeckhaus J, Weber LT, et al.:Protocol and rationale for a randomized controlled SGLT2 inhibitor trial in paediatric and young adult populations with chronic kidney disease: DOUBLE PRO-TECT Alport. Nephrol Dial Transplant. 2025, 40:679-87.
[4] Reiterová J, Tesař V. Current and Future Therapeutical Options in Alport Syndrome. Int J Mol Sci. 2023 Mar 14;24(6):5522.
[5] Kentarou Hashikami, Makoto Asahina, Kandai Nozu, Kazumoto Iijima, Michio Nagata, Michiyasu Takeyama,Establishment of X-linked Alport syndrome model mice with a Col4a5 R471X mutation,Biochemistry and Biophysics Reports,Volume 17,2019,Pages 81-86.

在线咨询


请添加“业务小助手”企业微信


电话咨询

业务咨询
中国:
+86-130-0328-0039(业务咨询)
+86(21)6176-5100(总机)
Email:info@pharmalegacy.com
海外:
+1-617-803-9415(海外业务咨询)
+86-021-6176-5100*8051(海外业务咨询)
Email:info@pharmalegacy.com
投资者:
+86(21)6176-5100*8002
Email:IR@pharmalegacy.com

TOP






 返回列表
返回列表










