


2026-03-16
模型相关
核酸药物凭借靶向致病基因、突破传统小分子与抗体药物不可成药靶点的颠覆性优势,已成为全球新药研发的核心赛道。2025 年,诺华 siRNA 药物 Leqvio 全球销售额突破 11.98 亿美元,标志着核酸药物正式迈入 “重磅炸弹” 行列;截至 2026 年 2 月,全球已上市核酸药物 20 余款,适应症从罕见病快速拓展至心血管代谢、神经退行性疾病等慢病领域,肝外递送技术更推动其向中枢神经系统(CNS)、肌肉等难治部位突破。
然而,核酸分子作为外源性生物大分子,其非预期免疫原性始终是临床开发与商业化应用的核心瓶颈 —— 既可通过激活先天免疫引发输注反应、细胞因子风暴等急性安全事件,也可诱导抗药抗体(ADA)产生,干扰药代动力学(PK)与药效学(PD),甚至导致治疗失效。基于此,本文整合核酸药物背景与分类、免疫原性核心来源、上市药物临床实证与核心规律、全球监管要求与法规建议、全链条评估体系与检测方法,结合最新行业实践与合规标准,构建标准化评估框架,为药物研发、生物分析与合规申报提供参考。
适用范围
本文聚焦以基因表达调控、蛋白替代或靶标特异性结合为核心治疗目的的核酸药物,涵盖反义寡核苷酸(ASO)、小干扰 RNA(siRNA)、治疗性 mRNA、剪接转换寡核苷酸(SSO)、核酸适配体等品种;核酸疫苗以主动激活免疫应答为治疗终点,其免疫原性评估逻辑与治疗性核酸药物存在本质差异,不纳入本文讨论范畴。
一、核酸药物的核心背景与分类
本文聚焦的核酸药物是一类通过调控基因表达或编码功能性蛋白发挥治疗作用的核酸类药物,主要包括寡核苷酸药物与治疗性 mRNA 药物两大类。其中,寡核苷酸药物分子量通常约 7–15 kDa,兼具小分子药物化学合成可控、结构明确、质量控制相对便捷的优势;治疗性 mRNA 药物分子量更大,一般可达 300–1500 kDa 甚至更高,更接近生物大分子特征,多以 LNP 递送,更易被模式识别受体识别、激活天然免疫通路。其分子结构、化学修饰方式、递送系统选择,是决定免疫原性风险等级的三大核心要素,直接主导后续评估策略的制定。
目前临床应用最成熟、研究最广泛的核酸药物可分为五大核心类别,各类别因结构与递送特性不同,免疫原性基础风险差异显著,具体分类及核心特征如下:
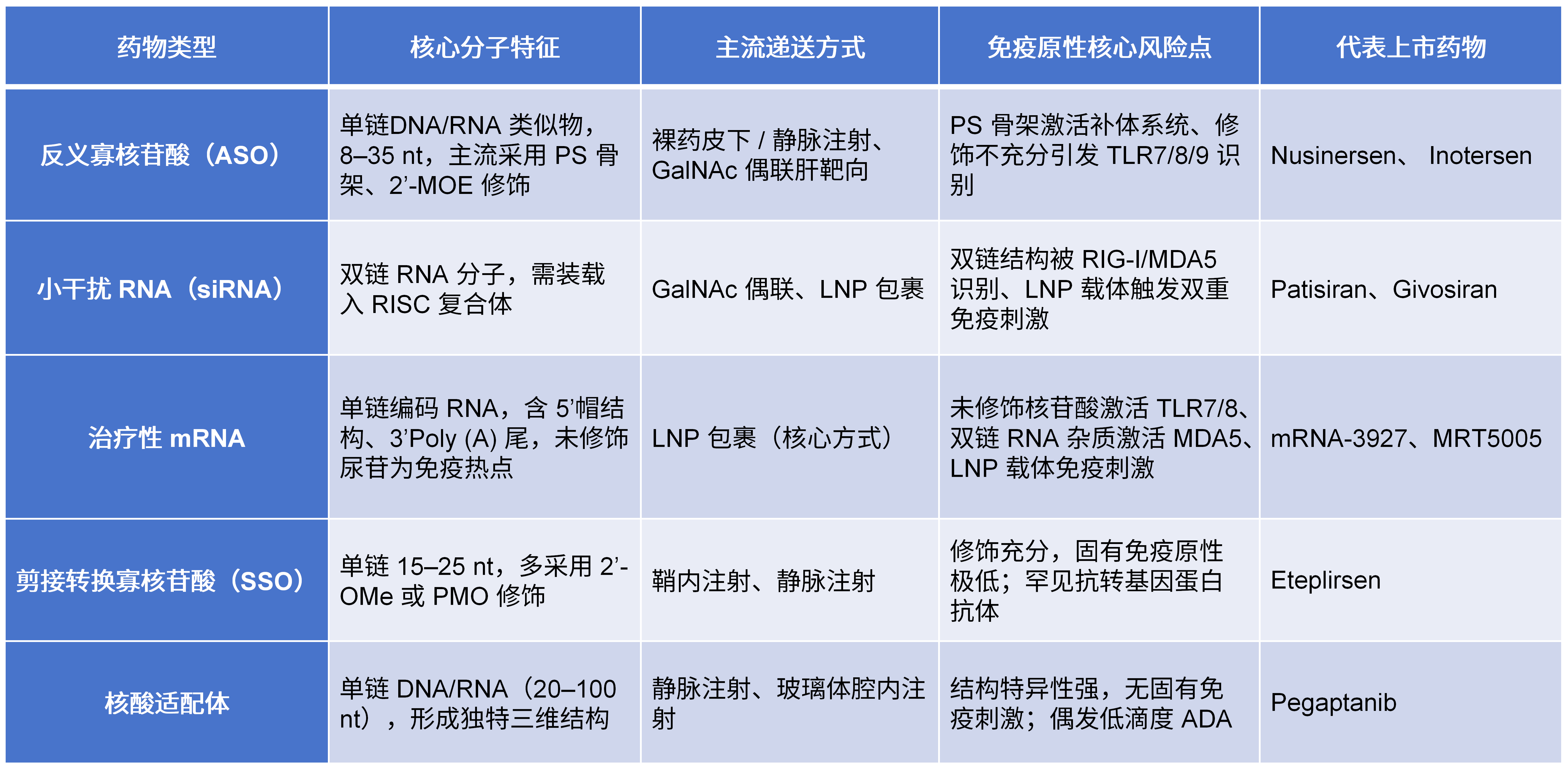
此外,微小 RNA(miRNA)模拟物 / 抑制剂、自扩增 RNA(saRNA)等新兴品类虽尚未大规模上市,但其免疫原性评估可遵循结构决定风险、递送放大风险的核心逻辑,参考上述五大类别的框架,结合自身序列特征、修饰方式与递送系统针对性调整。
二、核酸药物免疫原性的核心来源
与传统生物药(如抗体)主要激活适应性免疫不同,核酸药物的免疫原性源于先天免疫与适应性免疫的双重激活,且呈现先天免疫为急性风险核心、适应性免疫为长期风险关键的特征。核心风险因素可分为产品相关、工艺相关、患者相关、治疗相关四大类,其中产品相关因素起决定性作用,工艺与临床因素则为重要放大器。
1. 先天免疫激活:急性安全风险的首要诱因
先天免疫系统通过模式识别受体(PRRs)识别核酸药物中的病原相关分子模式(PAMPs),是输注反应、细胞因子风暴、补体激活相关伪过敏反应(CARPA)的核心诱因。其激活途径集中在核酸分子、递送系统、补体系统三个层面,且不同核酸药物的激活通路具有显著特异性。
1.1 核酸分子的固有免疫刺激
未修饰的天然核苷酸结构是固有免疫识别的核心靶点,ASO 与 siRNA 的未甲基化 CpG 基序、富含 GU 序列及双链结构,可被内体/溶酶体膜定位的胞内型Toll 样受体TLR3、TLR7、TLR8 和 TLR9以及胞质 RNA 受体RIG‑I/MDA5识别,进而触发促炎细胞因子释放。治疗性 mRNA 的未修饰尿苷及体外转录残留的双链 RNA 杂质,是 TLR3 与 MDA5 的强效激活剂。临床前研究证实,2'-MOE/2'- 氟修饰(ASO/siRNA)、假尿苷 / N1 - 甲基假尿苷替换(mRNA),可使先天免疫激活水平降低 90% 以上,这也是上市品种免疫原性可控的核心原因。
1.2 递送系统的免疫放大作用
递送系统是解决核酸药物稳定性差、胞内递送效率低的关键载体,同时也是核酸药物免疫原性的主要外源性诱因,不同递送系统的风险等级差异显著。LNP 载体风险最高,其可电离脂质能激活 TLR2/TLR4 与补体系统,PEG 化脂质因全球 30%–70% 人群存在预存抗 PEG 抗体,可加速 LNP 清除、降低药效,甚至引发严重过敏,新一代快速脱落型 PEG 脂质(如 PEG2000-C-DMG)已通过缩短脂质在体内的滞留时间,大幅降低该风险;GalNAc 偶联物免疫原性极低,仅偶发轻度注射部位反应;裸药递送的 ASO,其 PS 骨架可直接激活补体替代途径,是其补体激活风险的核心来源,与注射部位反应密切相关。
1.3 补体系统的异常激活
补体激活是连接先天与适应性免疫的关键通路,也是静脉高剂量给药时的主要风险。ASO 的 PS 骨架与 2'-MOE 修饰可激活补体替代途径,释放 C3a、Bb 等补体片段引发血管炎症;LNP 制剂中可电离脂质与PEG 脂质是诱发补体激活的主要成分,可通过经典/替代途径激活补体,诱发非 IgE 介导的伪过敏反应 CARPA,临床可通过对乙酰氨基酚联合糖皮质激素进行预处理,抑制补体激活相关炎症与类过敏反应,显著降低急性输注相关不良反应。
2. 适应性免疫激活:长期疗效的潜在挑战
适应性免疫激活的核心表现为抗药抗体(ADA)产生与 T 细胞活化,其触发条件较先天免疫更为严格,但对药物长期应用的影响更持久,是核酸药物长期疗效监测的核心内容。
2.1 ADA 的诱导机制与临床特征
核酸药物的 ADA 并非靶向核酸序列本身,而是针对 PS、2'-MOE 等化学修饰基团,PEG、GalNAc 等递送系统成分,或治疗性 mRNA 编码的异源转基因蛋白,且具有类特异性 —— 同一化学亚类的不同序列可能存在交叉反应,这为 “通用阳性对照” 的制备提供了理论依据。已上市品种中,90% 以上的 ADA 为 IgG/IgM 混合型、低滴度一过性抗体,不影响 PK/PD 与疗效;高滴度 ADA 多与高剂量、高频次给药相关,且多数情况下不影响 PK/PD 与疗效。
2.2 T 细胞活化的核心诱因
T 细胞活化仅见于 LNP 包裹的治疗性 mRNA、表达异源蛋白的 SSO 等高风险品种,普通 ASO、siRNA 药物极少出现。核心触发因素包括生产过程中残留的质粒 DNA、内毒素等工艺杂质的佐剂效应,及 LNP 介导的抗原交叉呈递 —— 激活细胞毒性 CD8+ T 细胞,导致药物作用靶细胞被清除,是 mRNA 药物研发需重点规避的风险。
3. 其他关键影响因素
除药物本身特性外,工艺、患者、治疗方式三类因素会进一步放大核酸药物的免疫原性风险,是评估与管控中需重点关注的环节:
3.1 工艺相关杂质
未修饰核苷酸、空 LNP、残留质粒 DNA、内毒素等 “隐形诱因”,是生产工艺质控的核心靶点,也被 EMA 2024 年指南明确纳入关键质量属性(CQA)。
3.2 患者相关因素
自身免疫病患者、免疫功能亢进者、既往接受过核酸药物治疗的患者免疫风险显著升高;预存抗 PEG / 抗 GalNAc 抗体是临床入排筛选的重要指标,高滴度患者需提前排除。
3.3 治疗相关因素
给药途径的免疫原性风险排序为静脉输注>皮下注射>免疫豁免部位给药(鞘内、玻璃体腔内等);长期重复给药的风险远高于单次给药,是慢病治疗用核酸药物给药方案优化的核心考量。
三、核酸药物免疫原性临床实证与核心规律
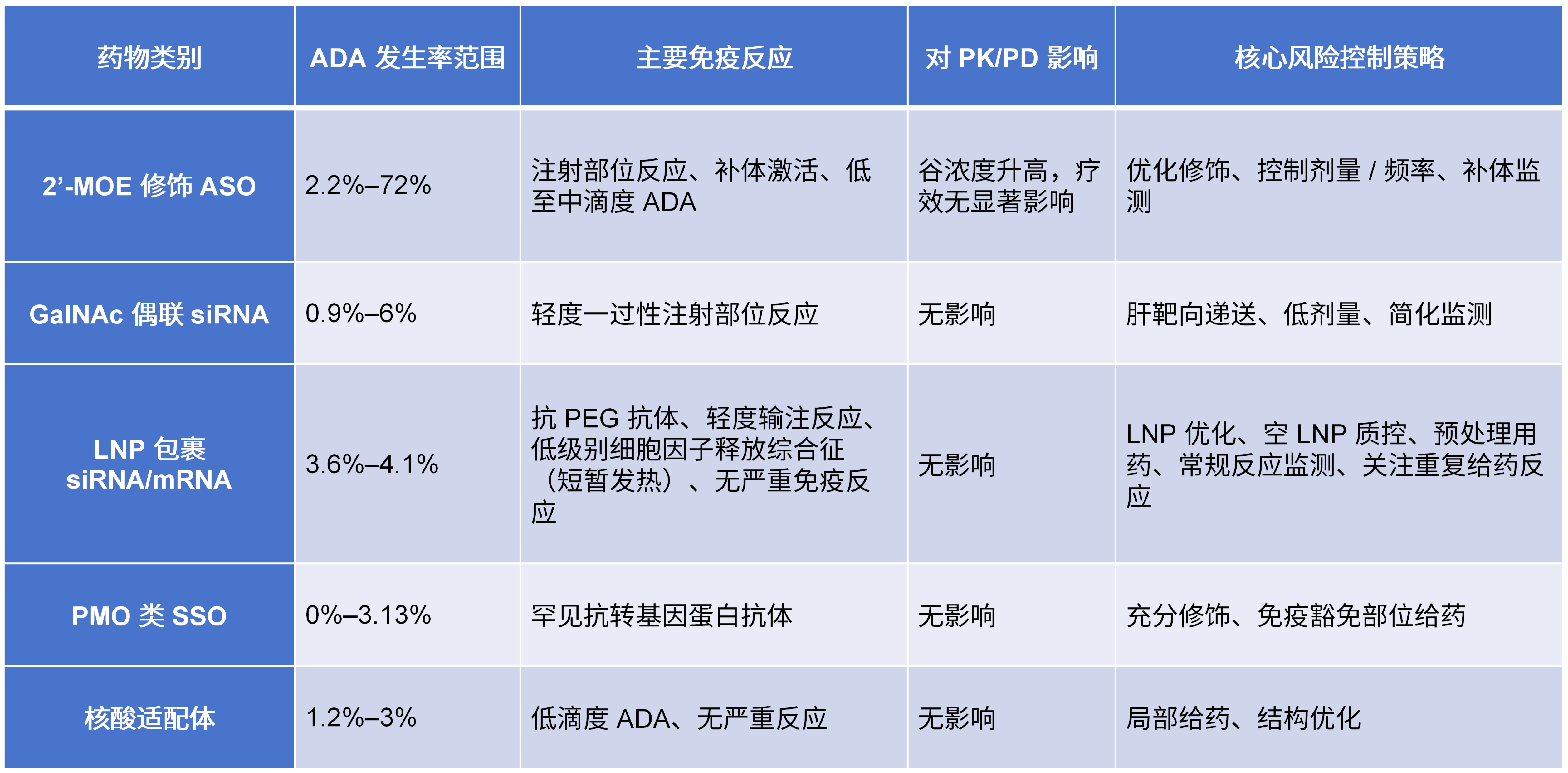
临床上核酸药物的免疫原性发生率呈现 0.9%–72% 的低至中等特征,且多数为轻度反应,对疗效与安全性无显著影响。这一结果得益于化学修饰、递送技术的持续优化,也为后续品种的免疫原性评估提供了风险分层、靶向管控的实证依据,6个典型临床案例的核心启示如下:
1. Patisiran(LNP 包裹 siRNA)
仅 4.1% 患者出现一过性低滴度抗 PEG 抗体,无抗 siRNA ADA 及严重 CARPA,ADA 对 PK/PD 及疗效无影响,提示LNP 成分精准优化是管控 siRNA 免疫原性的核心,优化后可采用简化监测策略。
2.Mipomersen(2'-MOE 修饰 ASO)
72% 患者出现 ADA(35% 高滴度),伴随补体 C3 消耗及注射部位反应,提示高剂量、高频次是 ADA 高发的主要诱因,需强化补体指标监测与注射部位反应管理。
3. Nusinersen(鞘内注射 ASO)
仅 4%–6% 患者出现一过性低滴度 ADA,无细胞因子激活与补体消耗,免疫相关不良反应接近 0,提示免疫豁免部位局部给药可显著降低免疫风险,低风险品种可大幅简化监测。
4. Givosiran(GalNAc 偶联 siRNA)
仅 0.9% 患者出现一过性低滴度 ADA,无严重过敏,提示GalNAc 偶联是 siRNA 免疫原性管控的 “金标准”,可采用 “样本保留 + 事件驱动检测” 策略。
5. Pegaptanib(核酸适配体)
ADA 发生率仅 1.2% 且滴度极低,无免疫相关严重不良反应,提示核酸适配体具有天然低免疫原性,结合局部给药可实现风险最小化,无需大规模监测。
6. HN2301(LNP 包裹治疗性mRNA)
3.8% 患者出现一过性低滴度抗 PEG 抗体,无抗 mRNA 特异性 ADA 及严重免疫相关不良反应,主要免疫反应为低级别细胞因子释放综合征(短暂发热),多在给药后 24 小时内自行缓解,重复给药后未出现反应加重现象,提示 LNP 配方优化(非炎性LNP)与 mRNA 核苷修饰的联合应用,可有效管控治疗性mRNA的免疫原性,对 PK/PD 及疾病治疗效果无影响,可采用常规反应监测、简化 ADA 监测策略,同时需关注重复给药后的免疫反应变化。
基于已上市品种的临床数据,不同类别核酸药物的免疫原性特征与管控策略可总结为:
四、核酸药物免疫原性评估的监管要求与法律建议
全球监管机构(FDA、EMA、NMPA、ICH)与行业组织(IQ 联盟、ISBA、AAPS)尚未出台针对核酸药物免疫原性评估的专属指南,但已形成介于小分子与生物制剂之间、基于风险的分层评估核心框架,核心依据为 ICH S6 (R1)、FDA 2024 年《寡核苷酸治疗药物临床药理学考虑》及 IQ 联盟 2025 年核酸药物免疫原性风险评估白皮书。
1. 核心监管文件及专属要求

2. 法规落地的核心实践建议
2.1 免疫原性风险评估(IRA)全周期动态实施
IRA 是评估策略制定的核心依据,需贯穿药物研发全流程。早期研发阶段基于序列特征、修饰程度、递送系统开展初步 IRA,筛选低免疫原性候选药物;临床前阶段结合人 PBMC 模型与非人类灵长类动物(NHP)实验细化 IRA,明确临床监测核心指标;临床阶段根据 Ⅰ/Ⅱ 期数据动态调整 Ⅲ 期策略,低风险品种简化监测,高风险品种强化 T 细胞活化评估与长期监测。
2.2 按风险等级实施分层监测
基于 IRA 结果将核酸药物分为低、中、高三个风险等级,实施差异化监测,完全对接监管 “风险导向” 要求:
低风险品种(GalNAc-siRNA、充分修饰 ASO、核酸适配体)仅收集保留样本,PK/PD 异常或出现安全信号时启动检测;
中风险品种(LNP-siRNA、部分修饰 ASO)实施先天免疫 + 适应性免疫双维度监测,检测细胞因子、补体与全流程 ADA;
高风险品种(LNP-mRNA、表达异源蛋白的 SSO)实施先天免疫 + 适应性免疫 + T 细胞活化全维度监测,监测周期延长至末次给药后 24 周。
2.3 标准化把控关键合规细节
临床入组前必须筛查抗 PEG、抗 GalNAc 预存抗体,高滴度患者(如抗 PEG≥1:100)需排除,排除标准需在临床方案中明确;共轭修饰类药物需开展域映射检测,精准区分不同靶点的 ADA;ADA 检测方法需完成全面验证,灵敏度要求 ASO/siRNA≤100 ng/mL、mRNA-LNP≤250 ng/mL,药物耐受性(DTL)需不低于治疗窗浓度的 10 倍;方法学开发阶段可引入酸解离、亲和捕获洗脱、固相萃取(SPE) 等样本预处理手段提升药物耐受水平,保障低滴度抗体精准检出;血清样本需在采集后 2 小时内分离,-80℃低温保存并避免反复冻融,建立全流程溯源体系。
五、核酸药物免疫原性的全链条评估体系与检测方法
基于全球监管要求与行业最佳实践,核酸药物的免疫原性评估已形成非临床早期筛选→临床前验证→临床全周期监测的全链条体系,同时围绕先天免疫、适应性免疫两大维度,建立了标准化、精细化的检测方法体系,确保评估结果的科学性、可重复性与临床相关性。
1. 全链条评估体系框架
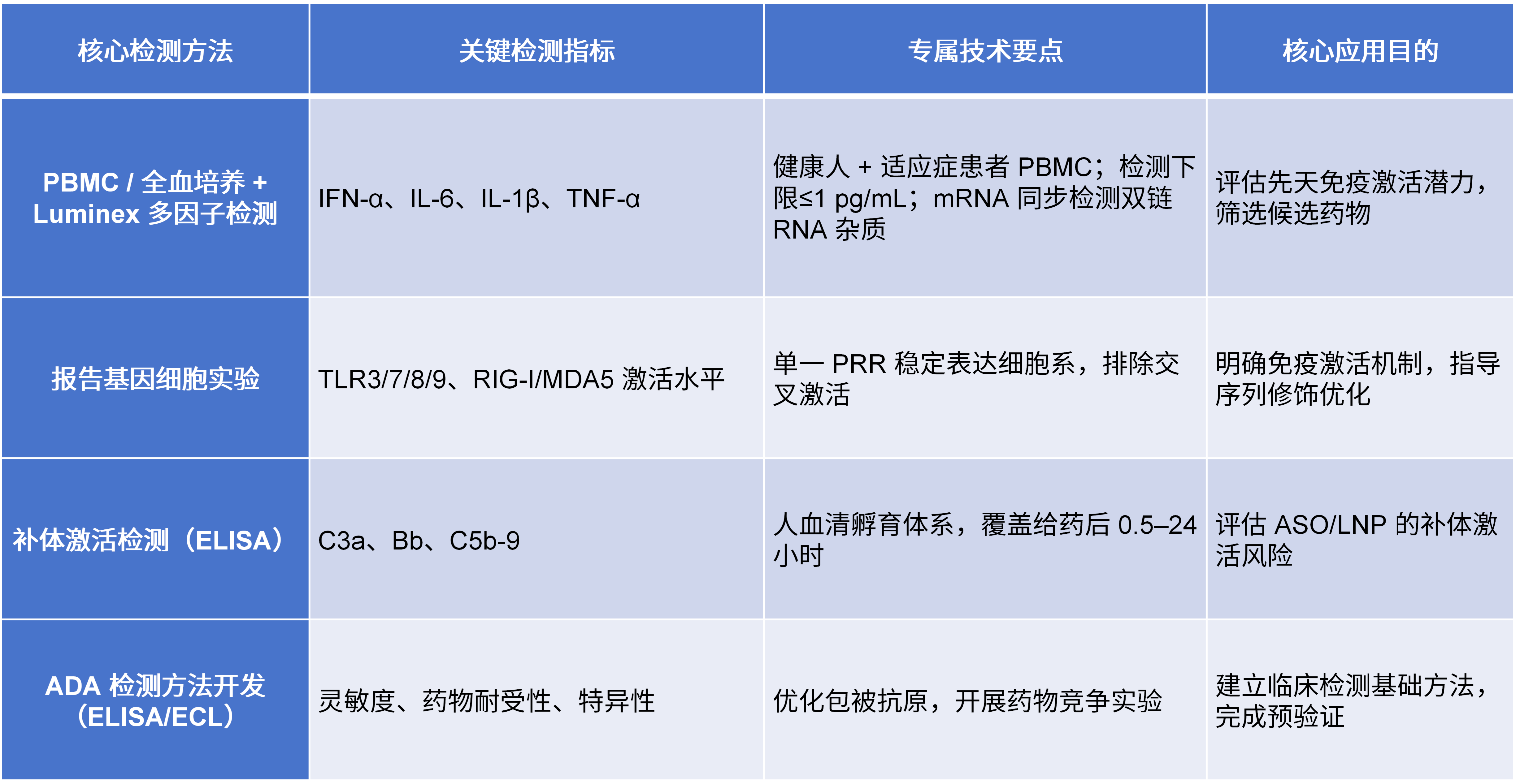
1.1 非临床评估阶段:源头筛选与风险预判
核心目标是筛选低免疫原性候选药物、优化产品设计与给药方案,严格遵循 ICH S6 (R1) 指南,分为体外与体内两大环节,其中体内评估环节通过NHP给药实验和疾病模型实验开展,具体为:NHP给药实验模拟临床给药方案,检测细胞因子谱、ADA滴度/亚型、组织免疫浸润,关联ADA与PK/PD/毒性以预测人体免疫反应、优化临床给药方案;疾病模型实验(SMA/血友病等疾病模型)检测免疫原性发生率、疗效保留率,验证疾病背景下的免疫风险。体外评估是基于体内样本进行的分析评估,具体细节如下:
1.2 临床评估阶段:全周期监测与风险评估
核心目标是监测免疫原性发生规律、评估临床相关性,贯穿 Ⅰ~Ⅳ 期临床试验,核心环节为样本采集与分层监测:样本采集遵循与 PK/PD 同步、覆盖关键时间点、兼顾事件驱动原则,给药前采集基线样本检测预存抗体,首次给药后 6h/24h 捕捉先天免疫峰值,每次给药前及末次给药后 4/12/24 周开展长期监测,高风险品种延长至 65 周;患者出现输注反应、过敏、PK/PD 异常时,立即采集样本开展应急检测。分层监测内容按风险等级精准实施,核心指标与检测频率严格匹配监管要求。
2. 核心检测方法的技术要点
2.1 先天免疫激活检测
以细胞因子、补体、PRR 激活为核心指标,细胞因子检测优先采用 Luminex 多因子平台,同步检测 15–20 种促炎 / 抗炎细胞因子,mRNA 药物可增加单细胞测序解析免疫细胞亚群活化变化;补体激活检测采用商业化 ELISA 试剂盒,检测 C3a、Bb、C5b-9 并覆盖给药后 0.5–24 小时,完整捕捉激活峰值;PRR 激活检测通过 qPCR 检测下游靶基因(IFN-β、MX1)表达,或通过报告基因实验特异性区分不同 PRRs 的激活水平。
2.2 适应性免疫激活检测
ADA 检测为核心项目,采用 ELISA / 电化学发光(ECL)桥联法,严格执行 “筛选→确证→滴度测定” 三层级流程,高风险品种需额外检测 IgG/IgM 亚型(IgM 为早期急性反应,IgG 为晚期持续反应);T 细胞活化检测为高风险品种专属,采用 ELISPOT 法检测抗原特异性 IFN-γ 分泌 T 细胞数量,流式细胞术检测 CD69、CD25 活化标志物并区分 CD4+/CD8 + 亚群,胞内细胞因子染色(ICS)区分 Th1/Th2/Th17 亚群,阐明免疫反应类型;中和抗体(NAb)评估采用间接评估法,对比 ADA 阳性 / 阴性患者的 PK 参数与 PD 标志物,分析 ADA 对药物暴露与药效的影响。
3.评估结果解读与全链条风险缓解策略
3.1 评估结果解读的核心原则
结果解读需坚持合规导向,遵循四大原则:
综合分析—— 结合 ADA 发生率 / 滴度 / 亚型、细胞因子 / 补体水平,关联患者免疫状态与给药方案全面评估;
临床相关性判定—— 短暂性低滴度 ADA(<1:16)且无免疫激活、PK/PD 异常者,判定为无临床意义;高滴度 ADA(≥1:128)、持续性存在且伴随安全信号者,重点评估风险;
因果判定—— 排除疾病、合并用药、感染等干扰因素,明确免疫原性与药物的直接因果关系;
动态评估—— 以 Ⅰ/Ⅱ 期数据优化 Ⅲ 期监测策略,以 Ⅳ 期数据开展长期监测,捕捉延迟性免疫反应。
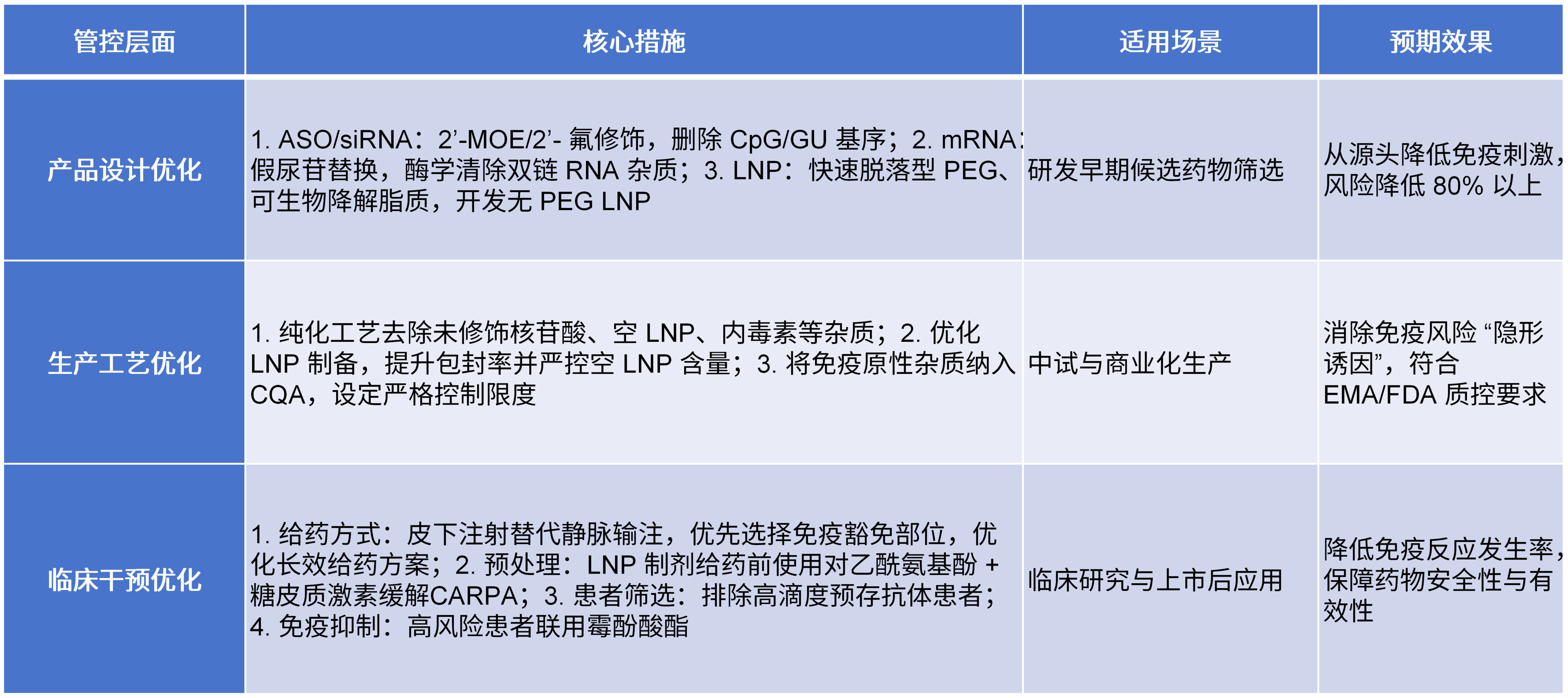
3.2 全链条风险缓解策略
从药物研发、生产到临床应用,构建多维度风险缓解体系,从源头降低免疫原性风险,保障药物安全性与有效性:
六、总结与展望
核酸药物的免疫原性评估已从早期 “单纯检测 ADA”,发展为基于风险、全链条覆盖、双维度监测、临床相关性导向的综合体系,其核心逻辑始终围绕 “在保障患者安全的前提下,加速核酸药物的临床转化”。当前,随着 2’-MOE 修饰、GalNAc 偶联、快速脱落型 PEG 等技术的成熟应用,多数上市核酸药物的免疫原性已实现 “低发生率、无显著临床影响”,为行业发展奠定了坚实基础。
同时,核酸药物免疫原性研究仍面临三大核心挑战:LNP 等递送系统的免疫激活机制尚未完全阐明;给药后 1 年以上的长期免疫原性数据积累不足;个性化免疫原性评估策略的落地难度较大。
未来,核酸药物的免疫原性评估将朝着精准化、智能化、个性化三大方向迈进:一是借助In Silico(计算机模拟)技术,整合序列、修饰、递送参数,精准预测免疫原性靶点,实现源头风险规避;二是将单细胞测序、质谱流式、空间转录组等新兴技术融入评估体系,更精准地解析免疫反应的细胞与分子机制,提升检测与评估效率;三是推动 FDA/EMA/NMPA 出台核酸药物免疫原性评估的专属指南,实现全球监管标准的统一与协调,加速核酸药物的研发与申报进程。
对于药物研发、生物分析与临床从业者而言,唯有紧密追踪全球监管动态,深耕核心技术研发,将免疫原性评估贯穿从候选药物筛选到上市后监测的全周期,通过科学的产品设计、严格的工艺控制与标准化的检测流程,才能持续推动核酸药物行业的高质量发展,让这一颠覆性疗法早日惠及更多患者。
澎立生物DMPK服务平台
澎立生物DMPK 服务平台,聚焦核酸药物研发全周期,依托成熟的生物分析体系、丰富的非临床 / 临床 PK/PD 研究经验,可实现核酸药物免疫原性与药代、药效数据的一体化解析,为药物免疫原性风险评估、给药方案优化、临床相关性判定提供精准数据支撑与定制化解决方案。平台可覆盖 ASO、siRNA、治疗性 mRNA 等全品类核酸药物,从早期候选药物筛选的免疫原性与 PK/PD 联合评价,到临床全周期的免疫原性 - PK/PD 动态关联分析,再到上市后风险监测的数据分析与策略优化,提供全链条、一体化的 DMPK 技术服务,助力药企高效破解核酸药物免疫原性研发痛点,加速药物临床转化与商业化进程。
未来,澎立生物将持续紧跟全球核酸药物研发与监管前沿,不断升级 DMPK 平台技术能力,深耕免疫原性与 PK/PD 的交叉研究领域,以专业的技术、标准化的体系、定制化的服务,与全球医药研发伙伴携手,共同推动核酸药物行业的高质量发展,让这一颠覆性疗法早日惠及更多患者。
参考文献:
[1] 2011 ICH Harmonised Tripartite Guideline Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals S6(R1)
[2] Anti-PEG immunity: emergence, characteristics, and unaddressed questions. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2015 Sep-Oct;7(5):655-77.
[3] The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv Drug Deliv Rev. 2016 Apr 1;99(Pt A):129-137.
[4] Immunogenicity for antisense oligonucleotides: a risk-based assessment. Bioanalysis. 2019 Nov;11(21):1913-1916.
[5] Studying how administration route and dose regulates antibody generation against LNPs for mRNA delivery with single-particle resolution. Mol Ther Methods Clin Dev. 2023 May 11;29:450-459.
[6] 2022 White Paper on Recent Issues in Bioanalysis: FDA Draft Guidance on Immunogenicity Information in Prescription Drug Labeling, LNP & Viral Vectors Therapeutics/Vaccines Immunogenicity, Prolongation Effect, ADA Affinity, Risk-based Approaches, NGS, qPCR, ddPCR Assays (Part 3 - Recommendations on Gene Therapy, Cell Therapy, Vaccines Immunogenicity & Technologies; Immunogenicity & Risk Assessment of Biotherapeutics and Novel Modalities; NAb Assays Integrated Approach). Bioanalysis. 2023 Jul;15(14):773-814.
[7] Considerations in the Immunogenicity Assessment Strategy for Oligonucleotide Therapeutics (ONTs). AAPS J. 2022 Aug 26;24(5):93.
[8] Assessment of the Immunogenicity Potential for Oligonucleotide-Based Drugs. Nucleic Acid Ther. 2022 Oct;32(5):369-377.
[9] Lipid nanoparticles for delivery of RNA therapeutics: Current status and the role of in vivo imaging. Theranostics. 2022 Oct 24;12(17):7509-7531.
[10] 2024 FDA Clinical Pharmacology Considerations for the Development of Oligonucleotide Therapeutics
[11] 2025 NMPA 先进治疗药品的范围、归类和释义(征求意见稿)
[12] Immunogenicity Risk Assessment for Nucleic Acid Therapeutics: A Comprehensive Evaluation for ASO, siRNA, and Nonvaccine mRNA/LNP Therapies by the IQ Consortium. Nucleic Acid Ther. 2025 Dec;35(6):261-279.
[13] 2025 EMA Guideline on the Development and Manufacture of Oligonucleotides

在线咨询


请添加“业务小助手”企业微信


电话咨询

业务咨询
中国:
+86-130-0328-0039(业务咨询)
+86(21)6176-5100(总机)
Email:info@pharmalegacy.com
海外:
+1-617-803-9415(海外业务咨询)
+86-021-6176-5100*8051(海外业务咨询)
Email:info@pharmalegacy.com
投资者:
+86(21)6176-5100*8002
Email:IR@pharmalegacy.com

TOP






 返回列表
返回列表










